B Cell Subtypes in Pre- and Postmenopausal Breast Cancer Patients

A creative scientist driven to solve biological problems and make meaningful contributions to research organizations.
A life sciences Ph.D. with expertise in diverse data modalities such as single-cell RNA sequencing, spatial transcriptomics, and imaging. My strong programming and data science skill sets allow me to solve complex problems efficiently. With experience in gene expression analysis, integrating omics datasets, and applying deep-learning techniques to NGS data, I can rapidly derive important insights from biological data sets.
As a former experimentalist turned computational scientist, I can capably collaborate with both computational and bench scientists alike. I’m creative, adaptable, and passionate about tackling challenges that will positively impact the lives of patients.
Swiss Institute of Bioinformatics
Data Scientist - -
Worked alongside the Translatioanl Data Science group I utlized data science, and bioinformatics to examine patient derived datasets to extract valuable biological insights.
Friedrich Miescher Institute (Novartis)
Computational and Experimental Biologist - -
Systems neuroscientist examining the relationship between computational models of neocortical function and transcriptionally defined cell types.
Click on the images below to learn more.
B Cell Subtypes in Pre- and Postmenopausal Breast Cancer Patients
Connecting computational roles to the transcriptome within neuronal cell types

Neural activity patterns of cortical subtypes in awake behaving animals

Characterizing cell type specific AAV Expressions Patterns
A Python Package for Predictive Modeling of Gene Expression Datasets

Home Office Drug Design: an ongoing newsletter & personal project


Brandeis University
Neuroscience Ph.D. -
Worcester Polytechnic Institute
Molecular Biology and Biotechnology -
Publications
Patent

Spatial transcriptomics project
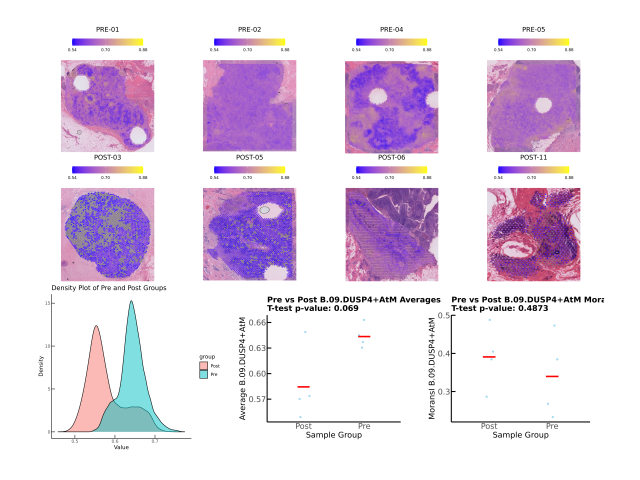
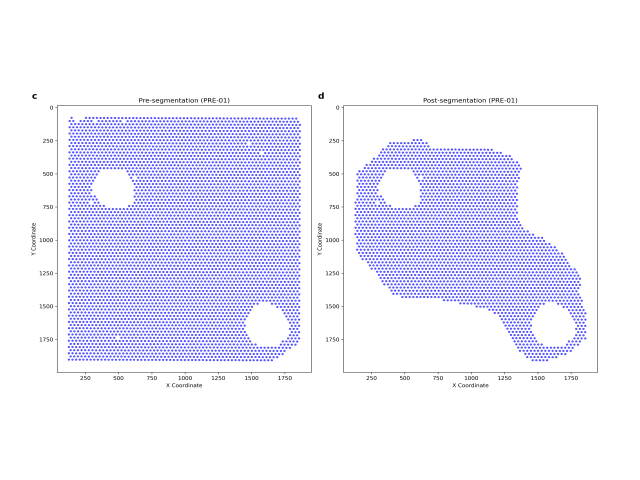
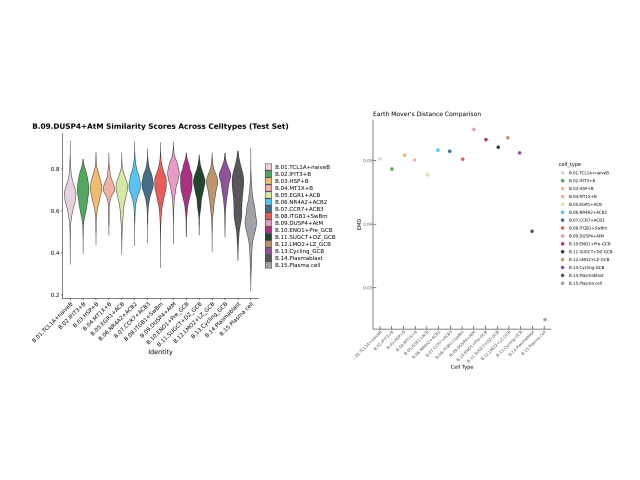
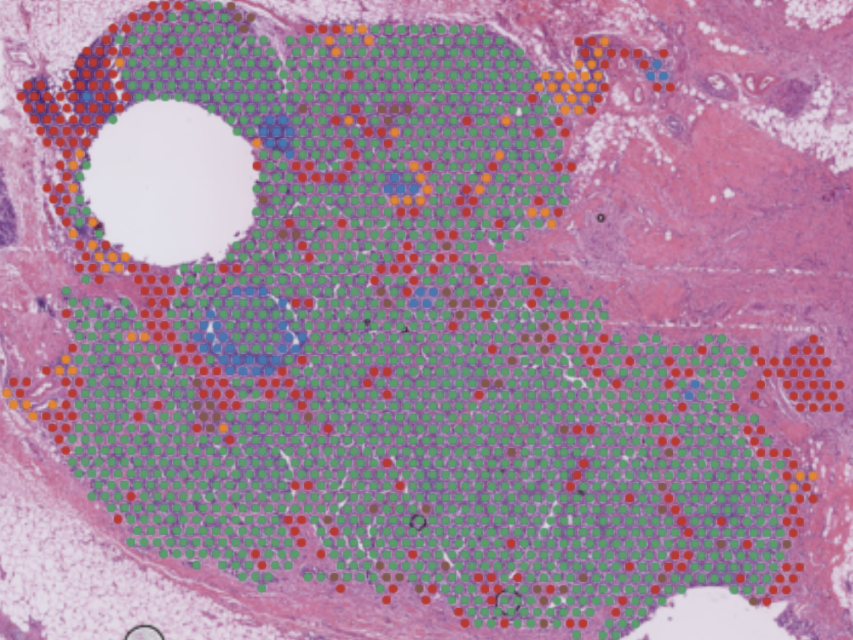
Question: Premenopausal women are at greater risk of experiencing more aggressive forms of breast cancer, often associated with poorer clinical outcomes compared to postmenopausal women. One potential explanation is differences in the composition of B cell subtypes among the immune cells infiltrating the tumor. In this project, I collaborated with physicians and analyzed patient-derived datasets to investigate the role of B cell subtypes in breast cancer samples from premenopausal and postmenopausal women.
Method: To determine whether B cell subtypes contributed to differences between these patient groups, I utilized spatial transcriptomics datasets derived from breast cancer patients and integrated them with externally sourced datasets. I identified cell type variations through automated cell type annotation and examined the cosine similarity scores between the samples of interest and a comprehensive B cell dataset.
Outcome: This work suggested that B cell subtypes vary meaningfully between these populations, providing insights into why these patients exhibit differing tumor progressions. The conclusions I drew were further validated using a separate bulk RNA sequencing dataset. The results of this analysis will be presented in a forthcoming medical paper.
Full report located hereSingle-cell RNA-seq Project
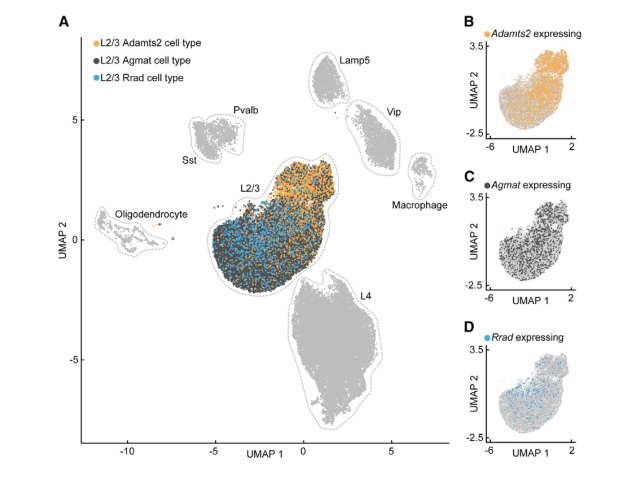
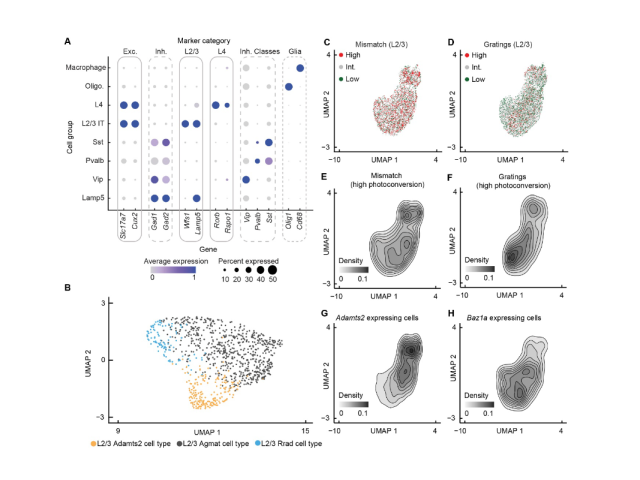
Question: A fundamental question in systems neuroscience is how the brain processes information. One prominent theory suggests that the brain uses prediction errors to update its own internal representation of the world. These prediction errors can be either negative (something expected is missing) or positive (something surprising happened), potentially corresponding to two distinct neuronal cell types. Here I set out to ask these functional cell types correspond to transcriptionally defined cortical subtypes.
Method:To accomplish this, I utilized single-cell RNA sequencing, optogenetics, virtual reality, and FACS to identify biomarkers associated with negative and positive prediction error neurons. My findings demonstrated that these neurons are transcriptionally defined cell types.
Outcome:This work enabled the development of cell-type-specific AAVs for labeling and studying these distinct neuronal populations, advancing our understanding of predictive coding in the brain.
Relevant publication hereNeural activity project
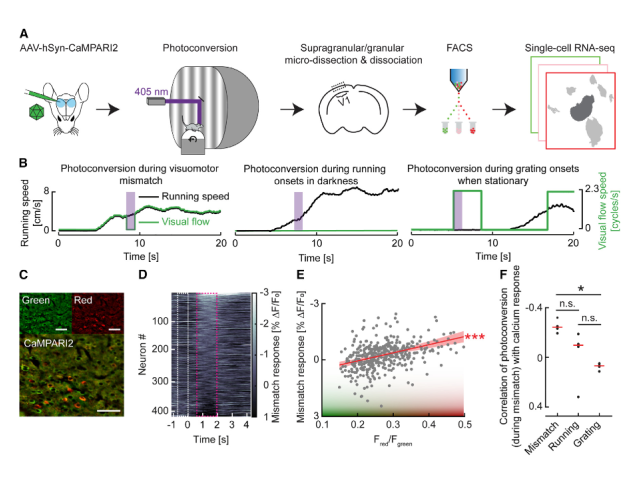
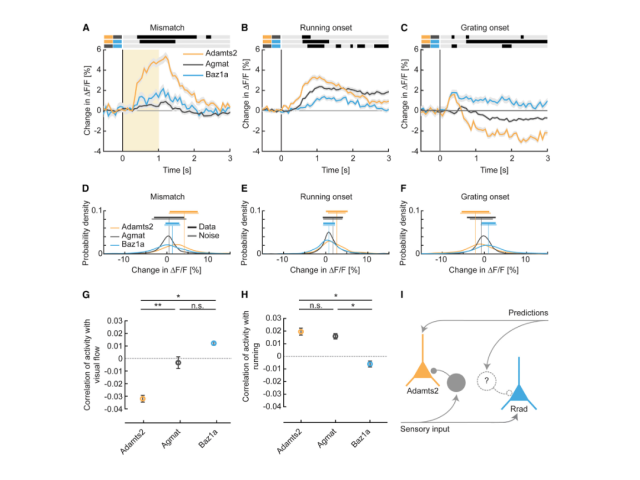
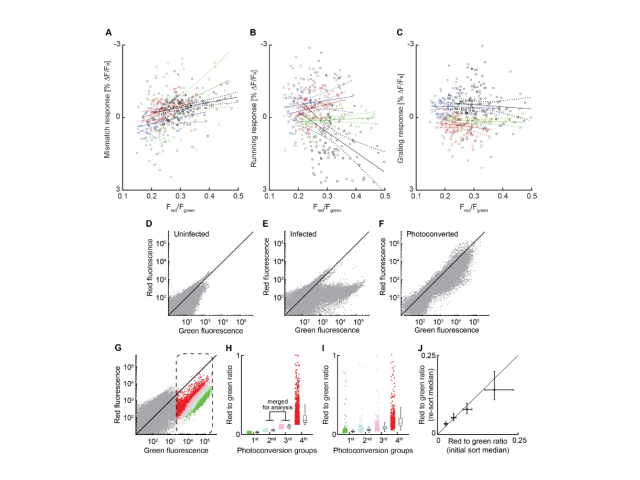
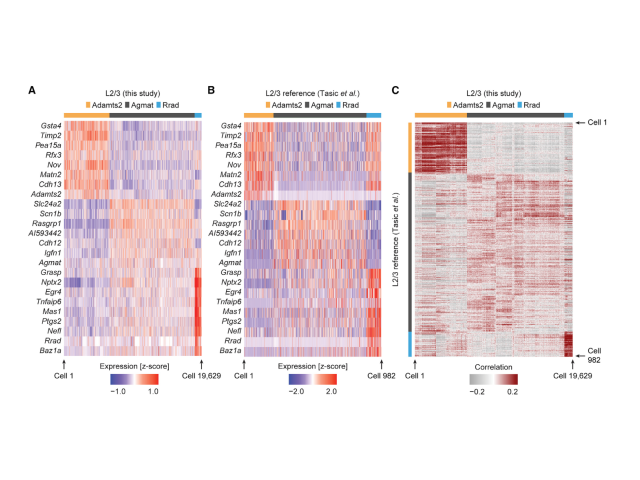
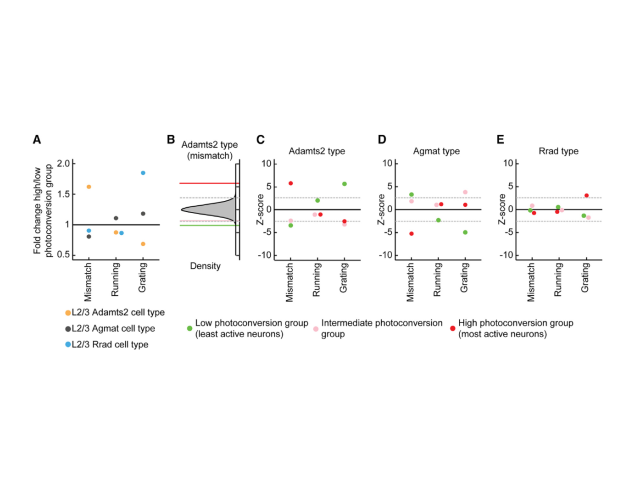
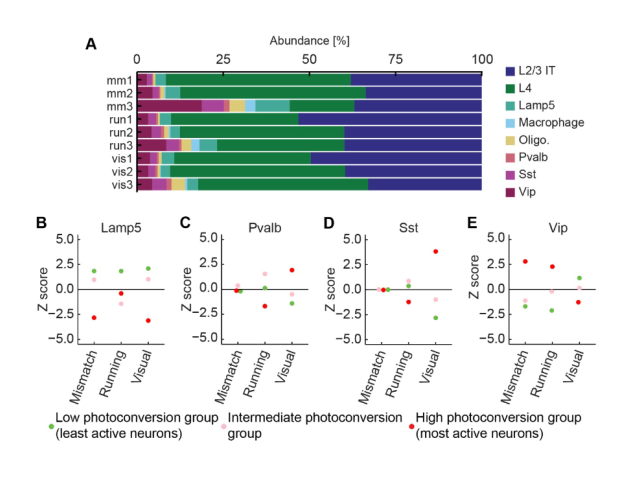
Question: Numerous studies have identified neuronal subtypes using dimensionality reduction and clustering of single-cell RNA sequencing datasets. However, the functional roles of these subtypes often remain unclear unless we can label and observe them in vivo. In this project, I investigated whether specific neuronal subtypes play distinct functional roles within the cortical circuit.
Method: To assess the functional specificity of several layer 2/3 subtypes labeled with AAVs employing biomarker-derived artificial promoters, I performed in vivo imaging of GCaMP-labeled neurons. Using two-photon calcium imaging, I observed neuronal activity while mice with cranial window implants navigated a virtual reality environment. I then analyzed the activity profiles of several thousand cortical neurons in MATLAB, employing time series analysis, regression, bootstrapping, and various statistical methods.
Outcome: This work demonstrated that cortical populations labeled with cell-type-specific AAVs exhibit distinct activity profiles, suggesting that the previously described neuronal subtypes in layer 2/3 represent transcriptionally and functionally distinct populations.
Relevant publication hereAAV characterization project
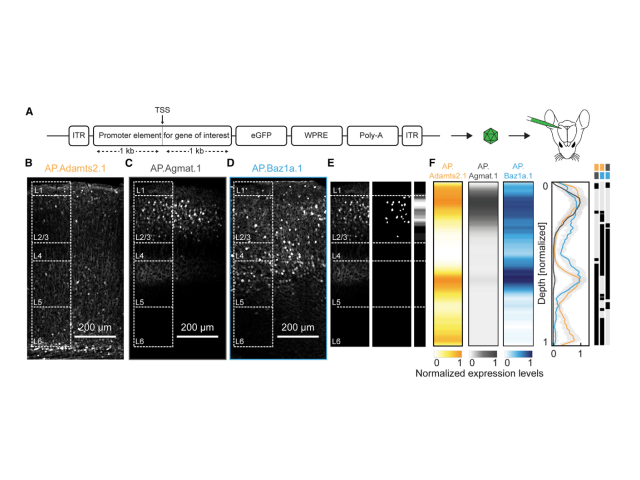
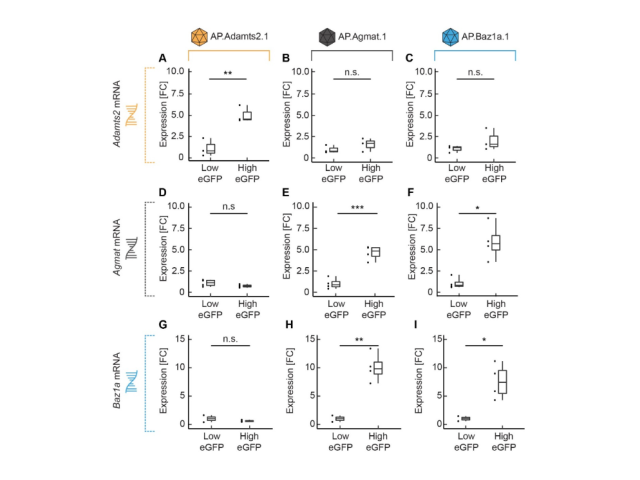
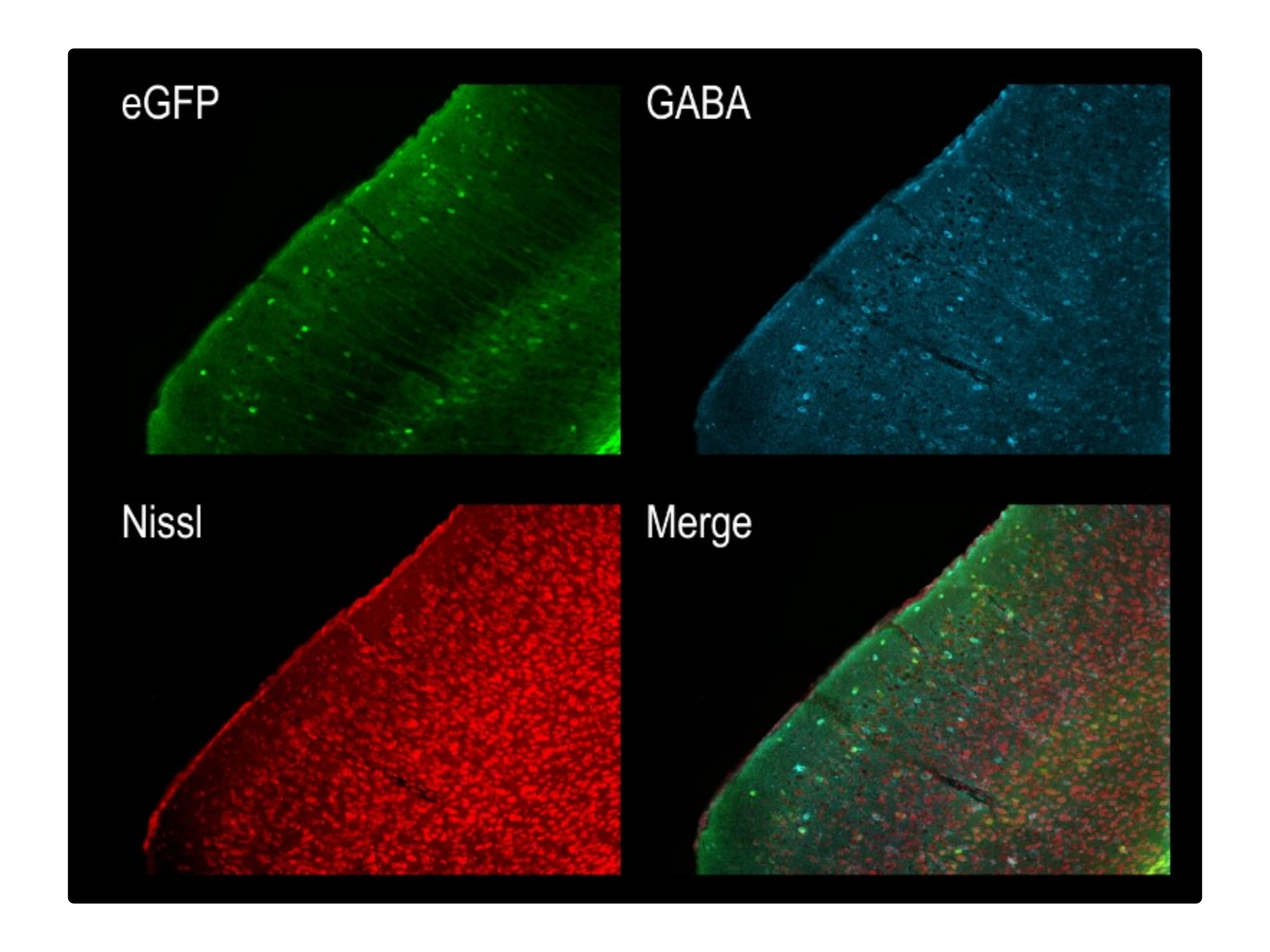
Question: We developed cell-type-specific AAVs based on biomarkers derived from single-cell RNA sequencing datasets. However, it was necessary to validate their specificity to ensure they were appropriate for functional validation of transcriptionally defined cell types. In this project, I collected and analyzed bulk RNA sequencing and histological datasets to test the specificity of the vectors we had designed, generated, and injected.
Method: To test vector specificity, I examined the differential expression patterns in R using FACS-derived bulk RNA sequencing datasets from animals infected with the cell-type-specific AAVs. Additionally, I analyzed the histological expression patterns of these AAVs using an image analysis pipeline I constructed in ImageJ and Python.
Outcome: This work demonstrated that the AAVs successfully labeled distinct populations and were specific enough to investigate their functional specificity.
Relevant publication herePredictive Analysis for Gene Expression
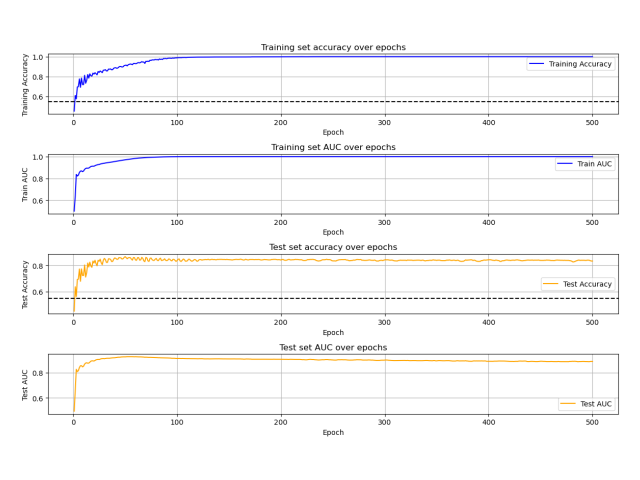
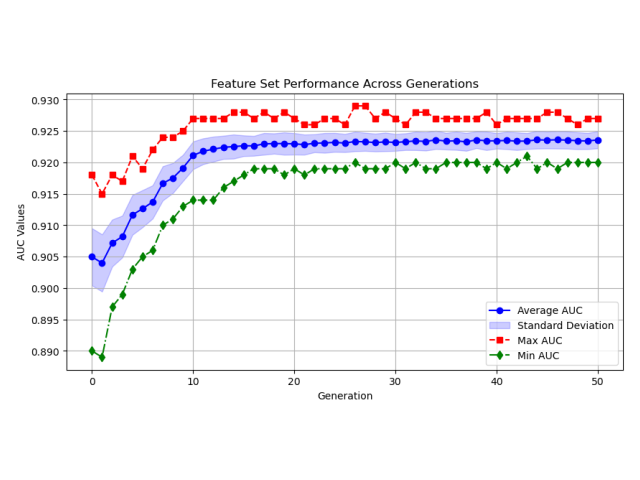
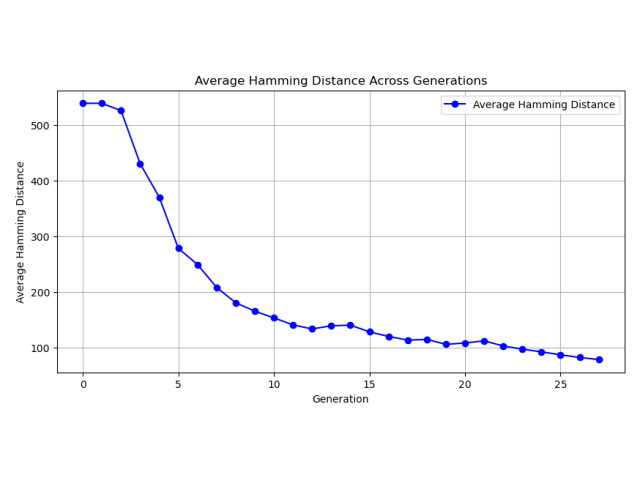
Goal: In my work with the Swiss Institute of Bioinformatics, I was frequently training neural to predict patient outcomes based on gene expression data sets. I was in need of a tool to rapidly test whether patient outcomes could be predicted from gene expression data. I developed a Python package that allows users to rapidly test whether gene expression data can be used to predict patient outcomes. The package is called PAGEpy (Patient Analysis Gene Expression Python).
Method: The packages integrates differential expression analysis, particle swarm optimization, data augmentation, batch normalization, and multi-layered neural networks to make prediction about a target variable based on gene expression data sets.
Outcome: The package is now available and can be easily installed with pip and is relatively user friendly. Please see the associated links below for more details.
Python Package
Github
repository
Personal newsletter project
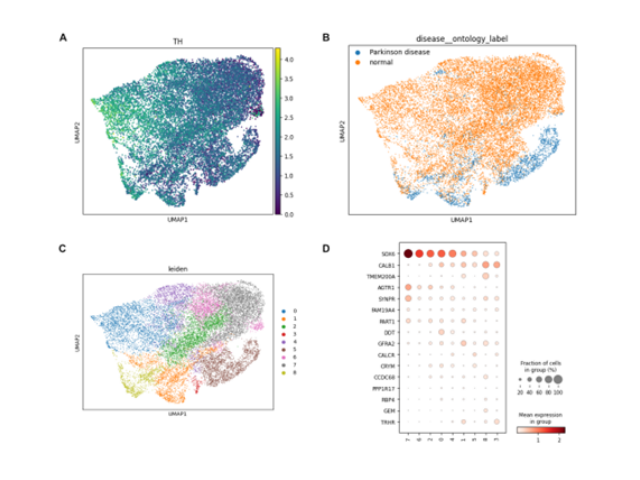
Summary: In this ongoing personal project, I am leveraging publicly available single-cell
datasets and foundation models to develop strategies for treating Parkinson's disease. The project is based
on the principle that a significant amount of biological research can be conducted computationally before
entering the lab. Feel free to subscribe to my newsletter or follow my GitHub
repository to stay updated on this work.
Check out the
newsletter
Relevant github repository